Essais cliniques – Profil d'innocuité
LUNESTA a généralement été bien toléré chez les adultes
Aperçu des effets indésirables du médicament1
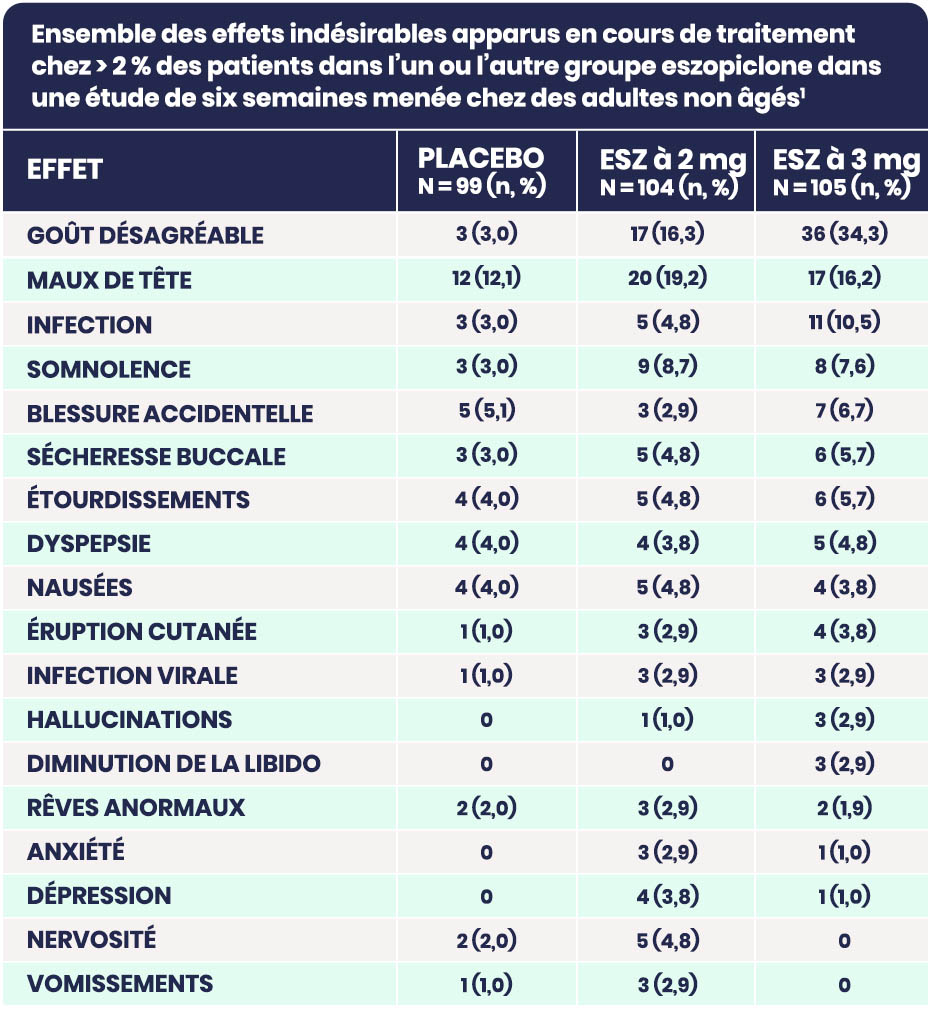
D’après la monographie du produit
Effets indésirables apparus en cours de traitement durant les essais cliniques contrôlés par placebo menés chez des adultes âgés de moins de 65 ans traités pendant une période pouvant aller jusqu’à 6 mois
Les types d’événements signalés étaient semblables à ceux observés dans l’essai de 6 semaines mené chez des adultes non âgés. Les autres effets indésirables signalés plus fréquemment avec l’eszopiclone qu’avec le placebo comprenaient les nausées (7,4 % p/r à 5,2 %) et la pharyngite (8,1 % p/r à 4,4 %)1.
Observations sur la tolérance dans les essais cliniques sur LUNESTA
- Dans les études cliniques sur LUNESTA, aucune tolérance à l’un des paramètres médians des mesures du sommeil n'a été observée pendant les périodes de traitement allant jusqu'à 12 mois1
- L’apparition d’une tolérance chez certains patients ne peut être exclue
Effets d'insomnie rebond avec LUNESTA
Une étude de six semaines menée chez des adultes a évalué l’insomnie rebond liée à LUNESTA (2 mg et 3 mg) les deux premières nuits suivant l’arrêt du traitement1,2
Avec les deux doses, 2 mg et 3 mg, l’effet émergent suivant l’arrêt du traitement :
- était léger
- présentait les caractéristiques du retour des symptômes de l’insomnie chronique
- semblait se résoudre la deuxième nuit suivant l’arrêt de LUNESTA
Dans le groupe LUNESTA à 2 mg, la DÉAE a augmenté, tandis que l’efficacité du sommeil a diminué, uniquement la première nuit, par rapport au début de la période de traitement.
La LAS persistant et la DÉAE ont augmenté de manière significative, tandis que l’efficacité du sommeil a diminué, la première nuit, par rapport au placebo. Aucune différence significative n’a été observée la deuxième nuit.
Dans le groupe LUNESTA à 3 mg, aucun changement par rapport au début de la période de traitement n’a été noté la première nuit.
L’efficacité du sommeil a significativement diminué la première nuit seulement par rapport au placebo.
La dose initiale recommandée est de 1 mg. La dose peut être augmentée à 2 mg ou 3 mg si cela est cliniquement indiqué. Il faut utiliser la dose efficace de LUNESTA la plus faible possible chez chaque patient.
Effets indésirables chez les patients présentant des affections médicales concomitantes
Trouble dépressif majeur (TDM) ou trouble d’anxiété généralisée (TAG)1
Dans deux études cliniques menées chez des adultes non âgés souffrant d’un TDM ou d’un TAG, couvrant un total de 8 semaines :
- Lorsque LUNESTA a été administré en association avec un inhibiteur sélectif du recaptage de la sérotonine, les incidences des EI ont été généralement semblables à celles liées au placebo
- Les EI qui ont été signalés plus fréquemment dans le groupe eszopiclone par rapport au groupe placebo comprenaient les suivants :
LUNESTA
Placebo
Asthénie
Sécheresse buccale
Somnolence
Étourdissements
Pharyngite
Goût désagréable
7,5 %
12,6 %
11,5 %
7,5 %
5,9 %
23,4 %
5,1 %
9,2 %
8,7 %
4,0 %
3,7 %
2,3 %
| LUNESTA | Placebo | |
|---|---|---|
|
Asthénie |
7,5 % |
5,1 % |
|
Sécheresse buccale |
12,6 % |
9,2 % |
|
Somnolence |
11,5 % |
8,7 % |
|
Étourdissements |
7,5 % |
4,0 % |
|
Pharyngite |
5,9 % |
3,7 % |
|
Goût |
23,4 % |
2,3 % |
Polyarthrite rhumatoïde1
Dans une étude clinique de quatre semaines menée chez des adultes non âgés atteints de polyarthrite rhumatoïde (PR) :
- Lorsque LUNESTA a été administré à des adultes atteints de polyarthrite rhumatoïde, les incidences des EI ont été généralement semblables à celles signalées dans l'essai clinique de 6 semaines
- Les EI signalés plus fréquemment dans le groupe eszopiclone par rapport au groupe placebo comprenaient l’asthénie (6,5 % p/r à 1,3 %), la pharyngite (5,2 % p/r à 0 %), et la polyarthrite rhumatoïde (18,2 % p/r à 9,2 %)
- Le traitement par l'eszopiclone à 3 mg n’a entraîné aucune aggravation de la PR sous-jacente chez ces sujets